Пожиттєвий діагноз. Що таке спінальна м’язова атрофія та чому її неможливо подолати

Спінальна м’язова атрофія (СМА) — дуже рідкісне захворювання, яке за рік в середньому діагностують у 35 українців. Однак почути абревіатуру СМА можна доволі часто: в інтернеті, на телебаченні та навіть у метро сьогодні нерідко трапляються повідомлення про збори коштів на дороговартісне лікування цієї хвороби. За так званий «укол життя», який може зупинити тяжкий розвиток діагнозу, треба викласти восьмизначну суму. Що варто знати про спінальну м’язову атрофію та чому в Україні пацієнти не можуть отримати допомогу від держави? Заборона поговорила про це з Віталієм Матюшенком, який очолює харківський благодійний фонд «Діти зі спінальною м’язовою атрофією».
Що таке СМА?
Спінальна м’язова атрофія — це спадкове генетичне захворювання, що вражає моторні нейрони у спинному мозку. Вони є керівними центрами організму та подають сигнали від мозку до м’язів і навпаки. За працездатність моторних нейронів відповідає білок SMN (survival motor neuron) — його продукує ген SMN1. Проте через мутацію в гені продукування білку зупиняється, а його відсутність провокує поступове відмирання моторних нейронів і слідом атрофію м’язів.
Захворювання передається від батьків до дитини за умови, якщо обидва мають зламаний ген. У такому випадку вірогідність його успадкування дорівнює 25%. Проявити себе СМА може в будь-якому віці — як у дитини, так і у дорослого, але який саме механізм є каталізатором цього процесу, науці наразі невідомо.
Залежно від проявів спінальної м’язової атрофії розрізняють чотири її типи. Для четвертого типу, який зазвичай виявляють у дорослих, характерні зовсім поверхневі прояви захворювання — зокрема, фізична втома. Для найважчого — першого — ураження м’язів дихання, ковтання та скелетної мускулатури. Такі пацієнти практично не доживають до чотирьох років і зазвичай помирають у перший рік життя.
Поширеність СМА суттєво не досліджена через її рідкісність. Це орфанна хвороба — тобто така, що трапляється не частіше ніж 1 випадок на 2000 населення. Згідно з загальносвітовою статистикою, один із 10 тисяч новонароджених матиме спінальну м’язову атрофію. В Україні, за даними Фонду хворих на СМА, 2020 року зареєстрували 45 випадків. Щорічна середня кількість нових реєстрацій за останні 3 роки в Україні становить 35 пацієнтів.
Як розпізнати захворювання?
Спінальна м’язова атрофія починається з м’язової слабкості та загальної втрати сил.
«Лікарі у переважній більшості кажуть: «Перечекайте, це вікове. Дитина просто не хоче рухатися, активно працювати. Бо їй комфортно так лежати. Бо вона наїлась. Бо вона просто така лінива. Не будемо нічого зараз робити. Згодом трошки активізується», — пояснює Віталій Матюшенко.
 Віталій Матюшенко
Віталій Матюшенко

Ця «лінивість» особливо проявляється в ранньому віці у найважчому типі СМА. Саме тоді потрібно негайно вдаватися до заходів з підтримки та убезпечення дитини від респіраторних захворювань, які у випадку ослаблення дихальних функцій можуть призвести до смерті.
У дитини самого Віталія Матюшенка ознакою захворювання стала так звана качина хода: «Це виставлений вперед животик, відставлена попка і рух трохи перевальцем, коли тулуп хитається й дитина перевалюється з ноги на ногу». Але такий прояв СМА можна помітити вже в тих пацієнтів, які досягли функції ходіння.
У дорослих захворювання не з’являється раптово: воно з ними впродовж усього життя. Проте характерні для спінальної м’язової атрофії ознаки не розпізнають на первинному огляді пацієнта, тож діагноз ставиться з великою затримкою.
Чи можна вилікувати СМА?
Генетичні захворювання не лікуються: змінити генетичну природу клітини вже неможливо. Її можна лише відкоригувати — або імітувати здоровий ген, або замістити ті функції, які ген не виконує.
На сьогодні є три лікарських засоби: «Золгенсма», «Еврісді» та «Спінраза». Перший спрямований на внесення в організм схожого здорового гену, який певний час буде заміщати роботу недієздатного гена. А два інших — на компенсацію недієздатності поламаного гена. Всі три в результаті сприяють продукуванню в організмі білку SMN, що захищає моторні нейрони.
Упаковка «Спінрази» – одного з препаратів, що полегшують життя пацієнтів зі СМА. Фото: Sebastian Gollnow/picture alliance via Getty Images

«Золгенсма» — це внутрішньовенна ін’єкція. Зробити її можна лише до конкретного моменту — поки дитині не виповниться 2 роки, а її вага не перевищить 21 кілограм. «Ми розраховуємо, що її дієвість, тобто те, скільки вона буде працювати, досягає п’яти років. Виробник гарантує 10 років. Але у будь-якому випадку після ін’єкції ми переступаємо межу летальності в два роки», — говорить Віталій Матюшенко.
«Спінраза» — теж ін’єкція, яку роблять в епідуральний (нижній) відділ спинного мозку. Впродовж першого року життя є навантажувальна доза, після чого треба пожиттєво робити укол тричі на рік. Натомість «Еврісді» — це сироп для використання в домашніх умовах. Його треба приймати пожиттєво, щодня в один і той самий час.
Таким чином вдається створити здоровий фон білка на ранній стадії, коли в організмі ще не почалися незворотні зміни. На більш пізніх стадіях хвороби можна гарантувати певну стабілізацію. І вже залежно від стану, до якого дійшов організм, можна говорити про ефективність реабілітації та відновлення втрачених функцій. Звісно, є певні функції, які неможливо відновити, тому що в організмі відсутні тканини, що мали б відповідати за їхнє виконання — наприклад, м’язові тканини за першого типу СМА.
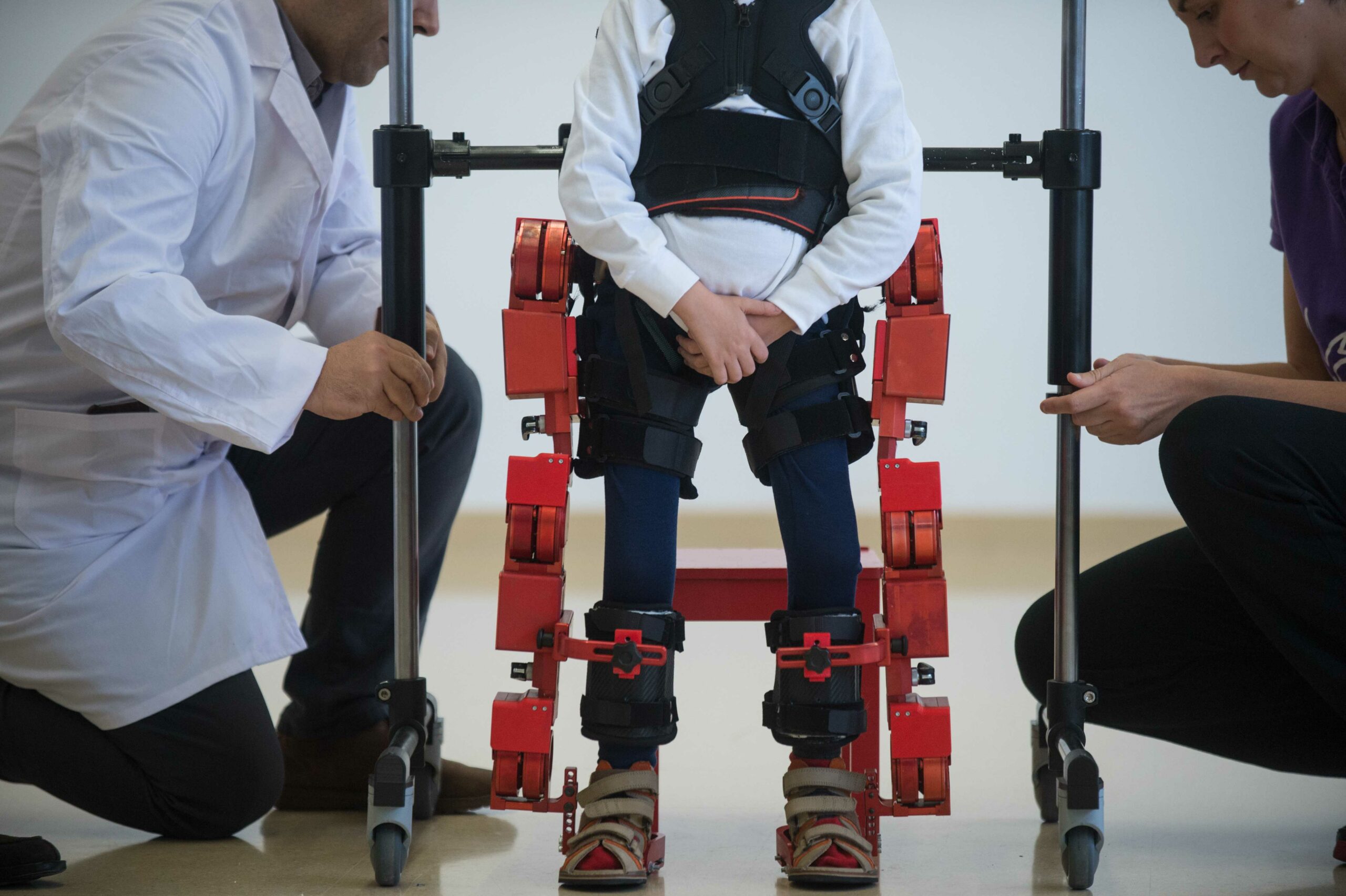
Йєнс, п’ятирічний хлопчик з діагнозом СМА ходить у своєму новому екзоскелеті, Барселона, 2017 рік. Фото: Lola Bou/Anadolu Agency/Getty Images
Наскільки ці ліки доступні?
Середня ціна «Еврісді» для України сягає близько 260 тисяч гривень. Тобто річна вартість сиропу у випадку максимальної дози складе близько 9 мільйонів гривень для одного пацієнта. Ін’єкція «Спінрази» в перший рік коштуватиме близько 12 мільйонів гривень, а в наступні роки — 5,5-6 мільйонів. Варість «Золгенсми» — 2,3 мільйона доларів. Купити препарат можна тільки в США.
Всі орфанні ліки є дороговартісними — не тому, що виробник хоче на них нажитися (хоча часто й через це також), а через високу вартість виробництва маленьких партій, оскільки ці препарати випускають рідко через недостатній попит.
Віталій Матюшенко зазначає, що такі ліки не розглядаються як ті, що можна придбати власним коштом: «Це апріорі прерогатива держави — забезпечувати таким лікуванням пацієнтів, які цього потребують. Це політика держави, політика системи охорони здоров’я, політика захисту своїх громадян».
В Україні наразі лікування від СМА ніяк не доступне, оскільки не існує жодної державної програми, яка фінансувала б придбання лікарських засобів і спеціального обладнання. Проте 2 червня 2021-го Верховна Рада України прийняла у першому читанні законопроєкт 4662, який визначає процедуру закупівлі дороговартісних інноваційних ліків за порядком укладення договору керованого доступу (ДКД).
У пояснювальній записці до законопроєкту говориться, що ДКД — це спеціальні договори між власником реєстраційного посвідчення на лікарський засіб та розпорядником бюджетних коштів. Такий договір укладається за результатами переговорів і дає змогу забезпечити пацієнтам доступ до інноваційних медичних технологій.
Віталій Матюшенко називає цей законопроєкт «лазівкою в українському законодавстві», оскільки він дозволить прописувати фінансування лікування окремим рядком у бюджеті без необхідності бути включеним у відповідну державну програму (якої наразі немає). Але поки невідомо, що буде після закінчення терміну ДКД. «Чи влада закриває проблему тимчасово, чи має якісь інші наміри, які можуть бути як корисними для пацієнтської спільноти, так і навпаки — обернутися нам ще гіршою ситуацією», — говорить Матюшенко.
Тобто єдиний вихід — це отримувати допомогу за кордоном?
Наразі так. Найближчий і найпростіший шлях — їхати до Польщі. Аби отримати допомогу, потрібно мати трудову візу й офіційне працевлаштування з оформленням державної страховки. А далі — звернутися до лікаря і підтвердити діагноз СМА. Для цього необхідно мати результати відповідного ДНК-тесту, який можна зробити і в Україні (Польща зазвичай додатково його не перевіряє).
«Найшвидше наше досягнення — місяць від перетину кордону до отримання лікування пацієнтом, — говорить Матюшенко. — Але якщо батьки до цього мали якісь трудові стосунки в Польщі, отримати допомогу можна буде ще швидше».


